La enfermedad de
Pompe es una enfermedad neuromuscular debilitante y rara que afecta a niños y
adultos. La forma infantil de la enfermedad se manifiesta generalmente en los
primeros meses de nacimiento, mientras que la forma juvenil tardía o adulta
aparece en cualquier momento durante la infancia o la edad adulta y tiene una
progresión más lenta. Ambos tipos de la enfermedad se caracterizan generalmente
por un debilitamiento muscular progresivo y dificultades respiratorias, pero la
gravedad de la enfermedad puede variar ampliamente dependiendo de la edad de
inicio y cuán afectados se encuentren los órganos. En la forma infantil, los
pacientes suelen manifiestar un corazón agrandado.
En niños:
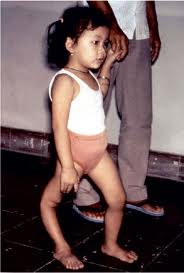
· Debilidad muscular progresiva.
· Tono muscular extremadamente disminuido.
· Lengua agrandada (y en algunos casos, protrusión de la lengua).
· Corazón agrandado y cardiomiopatía (enfermedad del músculo del corazón).
· Dificultad para respirar.
· Inhabilidad para alcanzar ciertas metas de desarrollo.
· Hígado agrandado (moderadamente).
· Dificultad para tragar, succionar, y/o alimentarse.
· Músculos faciales flácidos.
· Reflejos pobres o ausentes.

Tardía juvenil o adulta:
· Debilidad muscular progresiva, especialmente en el tronco y las extremidades inferiores.
· Fatiga al realizar esfuerzos.
· Dificultad progresiva para respirar.
· Dificultad para respirar al acostarse que mejora al sentarse o levantarse.
· Apnea del sueño (interrupción respiratoria temporal durante el sueño).
· Dolores de cabeza matutinos.
· Somnolencia y fatiga matutina.
· Tono muscular disminuido.
· Hígado agrandado.
· Lengua agrandada (poco común).
· Dificultad para masticar y tragar.
· Aumento en la frecuencia de infecciones respiratorias.
· Pérdida de reflejos.
· Dolor de espalda.
· Inhabilidad para alcanzar o pérdida de ciertas metas de desarrollo motor alcanzadas y niños.

Además de todo esto, si quereis más información sobre esta enfermedad, os recomendamos ver la película “Medidas extraordinarias” del año 2009, del director Tom Vaughan.